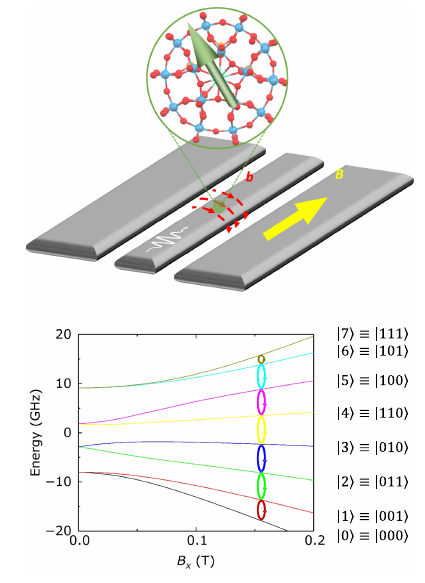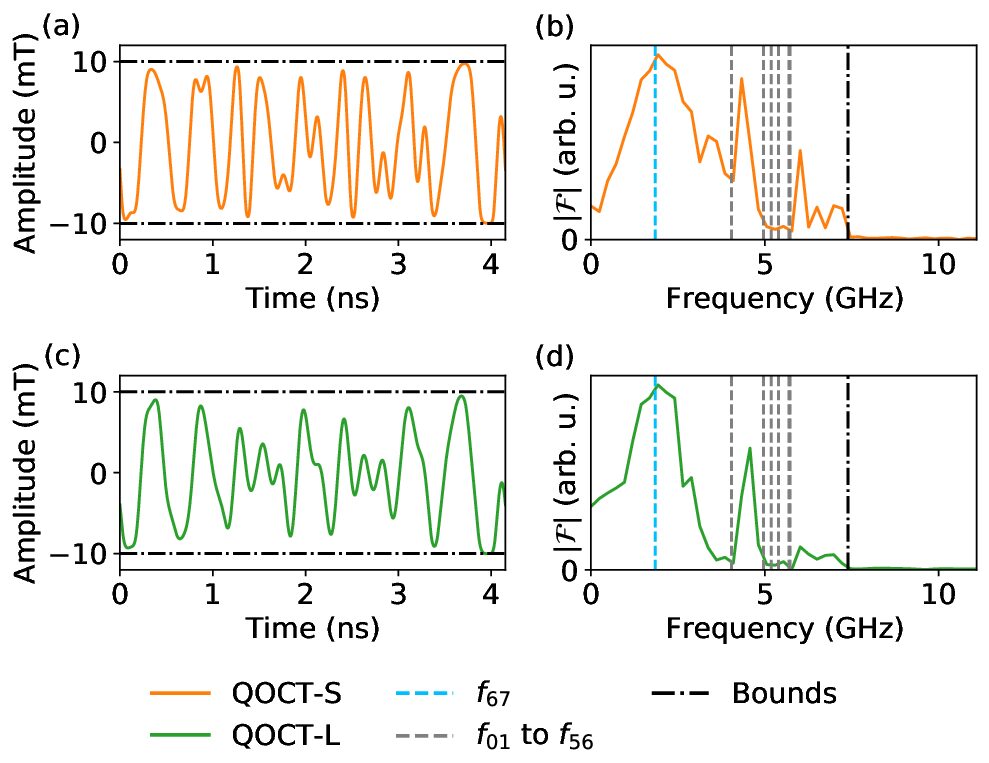
Alonso Hernández Antón, Fernando Luis and Alberto Castro, Quantum Science and Technology 10, 025042 (2025)
Quantum optimal control theory (QOCT) can be used to design the shape of electromagnetic pulses that implement operations on quantum devices. By using non-trivially shaped waveforms, gates can be made significantly faster than those built by concatenating monochromatic pulses. Recently, we applied this idea to the control of molecular spin qudits modeled with Schrödinger’s equation and showed it can speed up operations, helping mitigate the effects of decoherence (Castro et al 2022 Phys. Rev. Appl. 17 064028). However, short gate times require large optimal pulse amplitudes, which may not be experimentally accessible. Introducing bounds to the amplitudes then unavoidably leads to longer operation times, for which decoherence can no longer be neglected. Here, we study how to improve this procedure by applying QOCT on top of Lindblad’s equation, to design control pulses accounting for decoherence already in the optimization process. We define the control signal in terms of generic parameters, which permits the introduction of bounds and constraints. This is convenient, as amplitude and frequency limitations are inherent to waveform generators. The pulses that we obtain consistently enhance operation fidelities compared to those achieved with the optimization based on Schrödinger’s equation, demonstrating the flexibility and robustness of our method. The improvement is larger the shorter the spin coherence time T2.