Adrián Gómez Pueyo, Miguel A. L. Marques, Angel Rubio, and Alberto Castro, J. Chem. Thelr. Comp. 14, 3040 (2018)
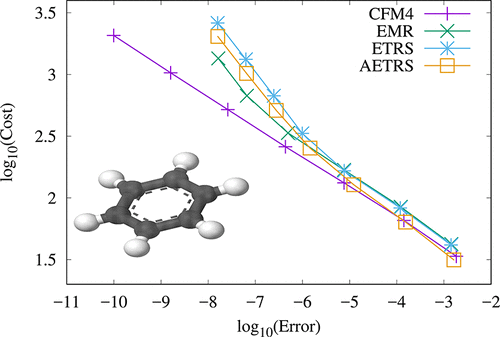
We examine various integration schemes for the time-dependent Kohn−Sham equations. Contrary to the time-dependent Schrödinger’s equation, this set of equations is nonlinear, due to the dependence of the Hamiltonian on the electronic density. We discuss some of their exact properties, and in particular their symplectic structure. Four different families of propagators are considered, specifically the linear multistep, Runge−Kutta, exponential Runge−Kutta, and the commutator-free Magnus schemes. These have been chosen because they have been largely ignored in the past for time-dependent electronic structure calculations. The performance is analyzed in terms of cost-versus-accuracy. The clear winner, in terms of robustness, simplicity, and efficiency is a simplified version of a fourth- order commutator-free Magnus integrator. However, in some specific cases, other propagators, such as some implicit versions of the multistep methods, may be useful.